每頁:左邊投影片、右邊中文講稿。手機自動改成上下排列。也可下載左圖右稿的對照 PDF離線看。

封面 · Cover
今天我想說服各位一件事:在 Pt-skin 高熵合金上,催化劑設計該瞄準的,不是單一活性位點,而是一整個「活性位點族群」。我是輔仁大學化學系的廖振成。這份工作會帶大家走一條線——埋在底下的成分控制電子結構、電子結構控制吸附、吸附決定整個活性位點族群;預測,只是這套框架自然的產物。今天記得一句話就好:我們要設計的,是那群埋在底下的原子。
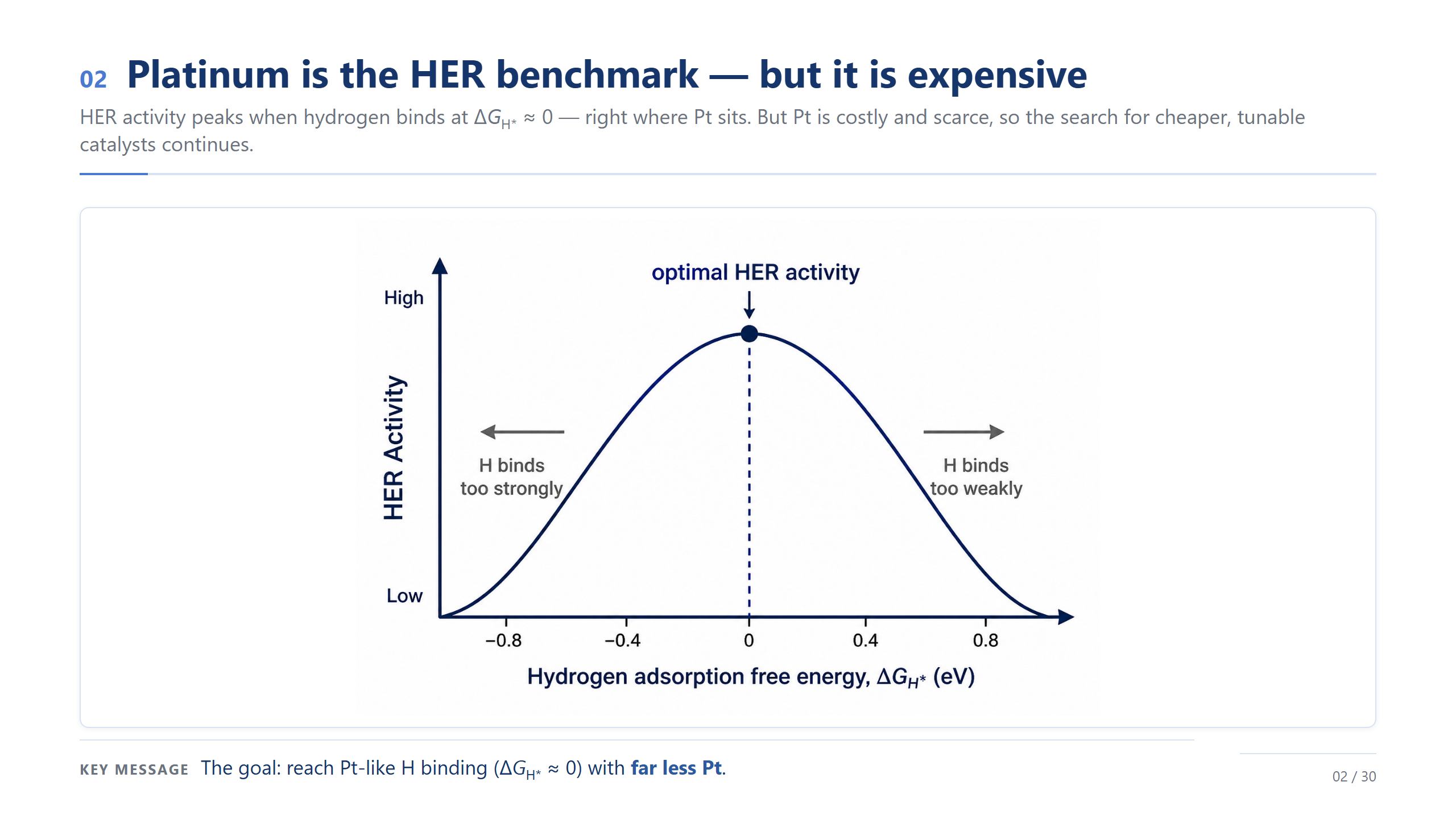
Pt 是基準,但很貴
故事從一個很實際的矛盾開始。HER 的活性由一個量決定:吸附氫的自由能 ΔG_H*,太強太弱都不好、接近零最理想,而白金正好坐在這個最佳點上。問題是 Pt 又貴又稀有。所以真正的挑戰不是「找到好催化劑」,而是「用更少的 Pt,達到跟 Pt 一樣接近零的氫吸附」。這就是我們整個設計的起點。

文獻漏斗:從 Pt-skin 合金設計到 active-site distribution
在講細節之前,我先用一頁把文獻的脈絡收成一條線。HER 催化的設計,通常從一個描述符出發:氫吸附自由能 ΔG_H*,不能太強也不能太弱。進到 Pt-skin 合金,問題就不只是表面是 Pt——底下的次表層成分會透過 ligand effect 和 strain effect 去調控表面 Pt 的吸附。而高熵合金又讓這件事更進一步:它不是只有一種理想位點,而是一整群不同 local environment 的集合。所以這份工作真正要問的,不是單一位點的 ΔG_H* 是多少,而是次表層的局部成分如何決定接近最佳那一群 active site 的比例。換句話說,Pt-skin 高熵合金的活性應該用統計的角度來理解:表面是一個位點族群,而埋在底下的成分,控制著 ΔG_H* 的分布。

高熵合金 = 巨大的局部環境多樣性
那替代方案是什麼?高熵合金。但今天我不打算花時間教熵——重點只有一個:高熵合金把五種以上的元素亂混在一起,於是表面上沒有兩個位點看到一樣的鄰居。它打開了一個龐大、可連續調整的成分空間。對催化來說,高熵合金真正迷人的地方,是一個材料裡就藏著無數種不同的局部環境、無數種不同的位點。
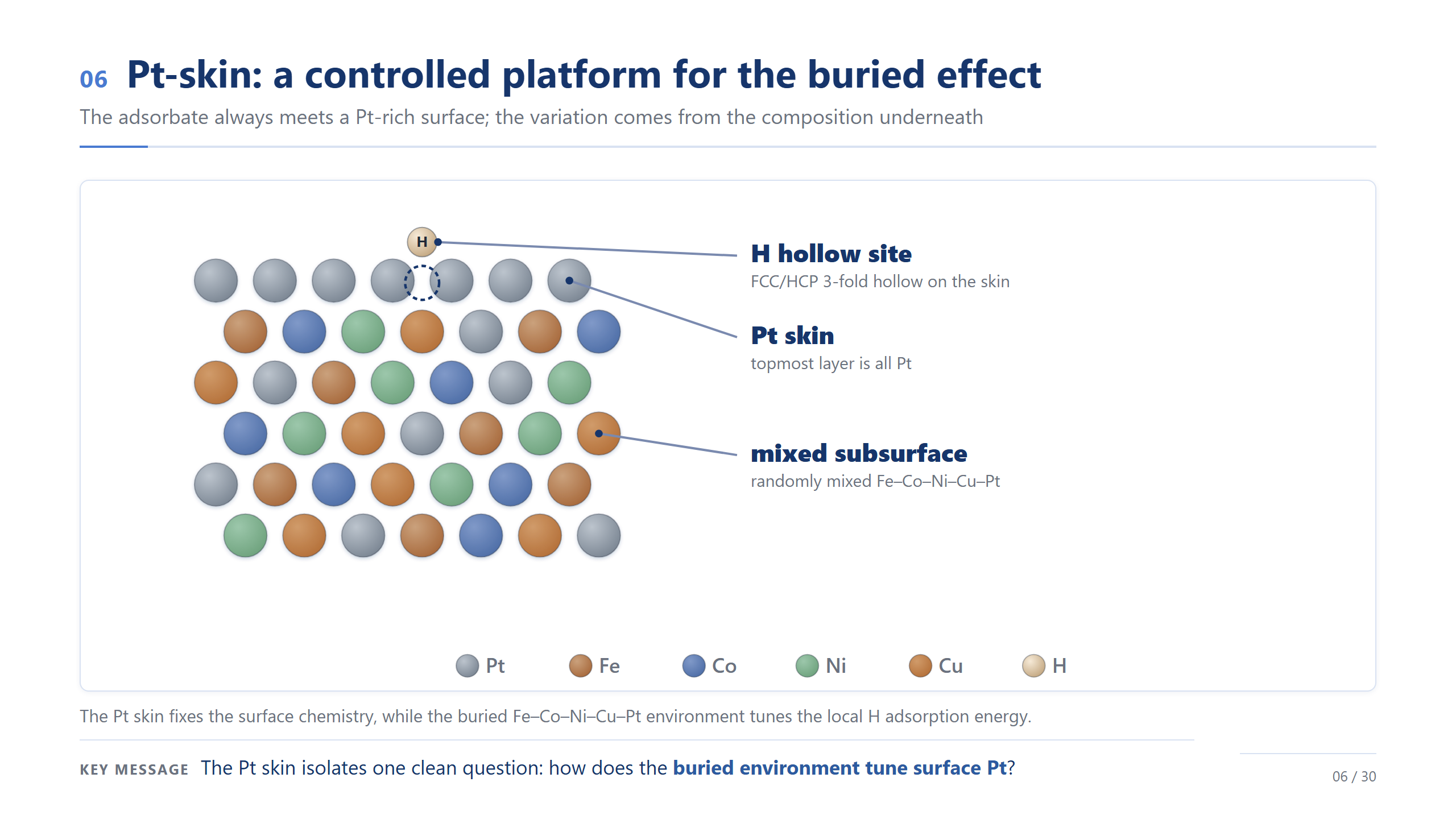
Pt-skin:保留表面、調控內層
那怎麼同時保留 Pt 的好、又拿到高熵合金的可調性?答案是 Pt-skin。最左邊純 Pt,表面好但貴;中間一般合金,連 Pt 的表面化學都不見了;最右邊是我們的設計——最上層全是 Pt、表面化學跟 Pt 一樣,底下卻藏著 Fe–Co–Ni–Cu–Pt 的混合。一句話:把 Pt 的表面留著,讓底下的原子去調它,而且用更少的 Pt。
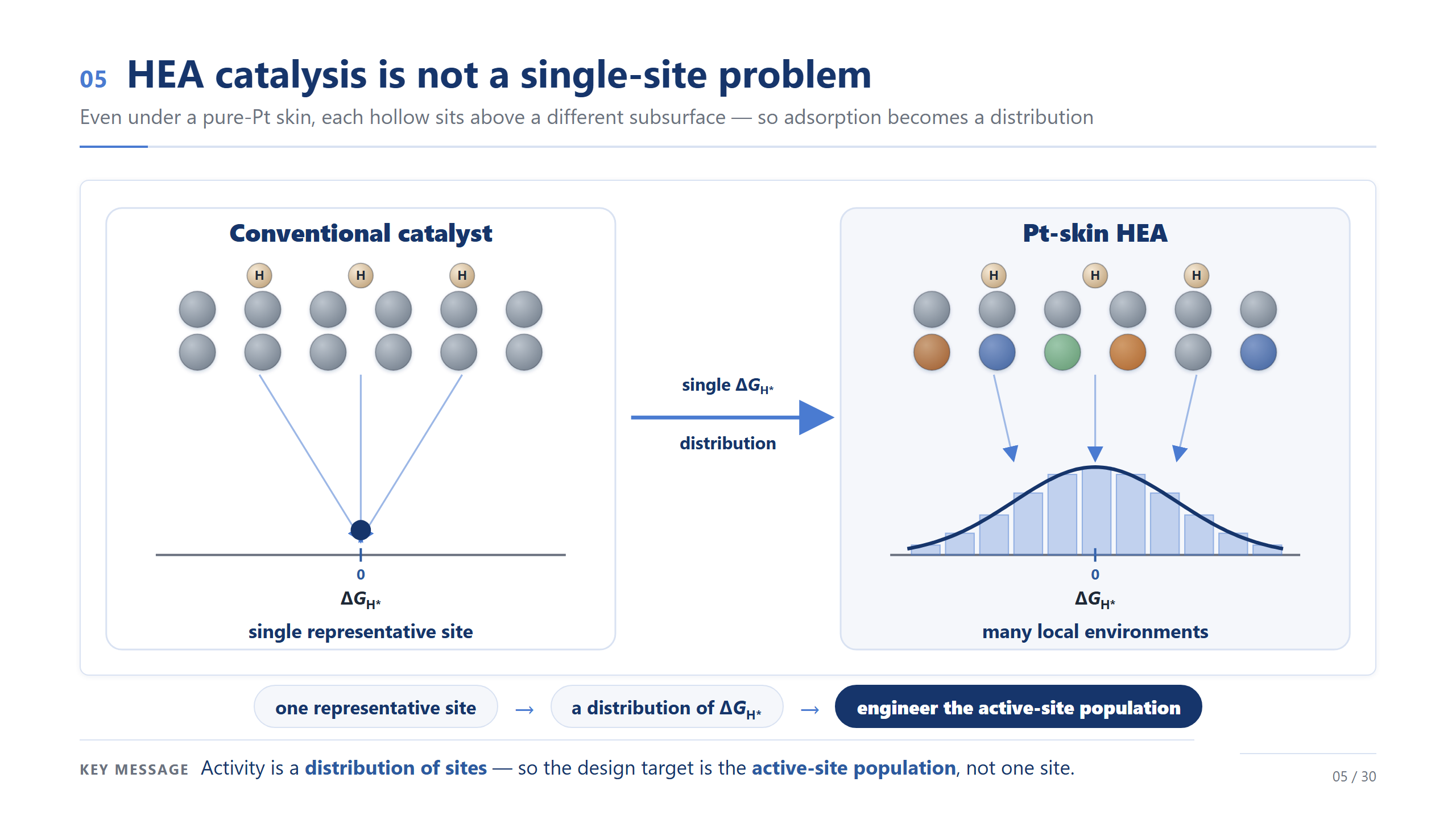
不是單一位點,是位點族群
這是今天最重要的觀念轉變,請特別記住這一頁。左邊就算是均勻的傳統表面,氫的吸附位也只有 top、bridge、hollow 這幾種——幾個離散的位、每種一個明確的 ΔG。但右邊的 Pt-skin 高熵合金,光是 hollow 這一種位,就因為底下環境每個都不一樣,而散成一整個 ΔG 的分布。從幾個離散的位、到一個連續的分布、到一個要去設計的族群:從這頁開始,請把催化想成一個活性位點族群的問題,而不是某一個位點。
Pt-skin 是受控平台
要研究「底下怎麼影響表面」,Pt-skin 剛好是一個乾淨的受控平台。注意:吸附的氫永遠落在 Pt 上,表面化學被釘死了,唯一在變的是底下的成分。所以這個系統幫我們把問題隔離成一個很單純的提問——在表面不變的前提下,底下的環境怎麼調表面 Pt?這正是我們能把「埋藏效應」單獨拉出來看的原因。
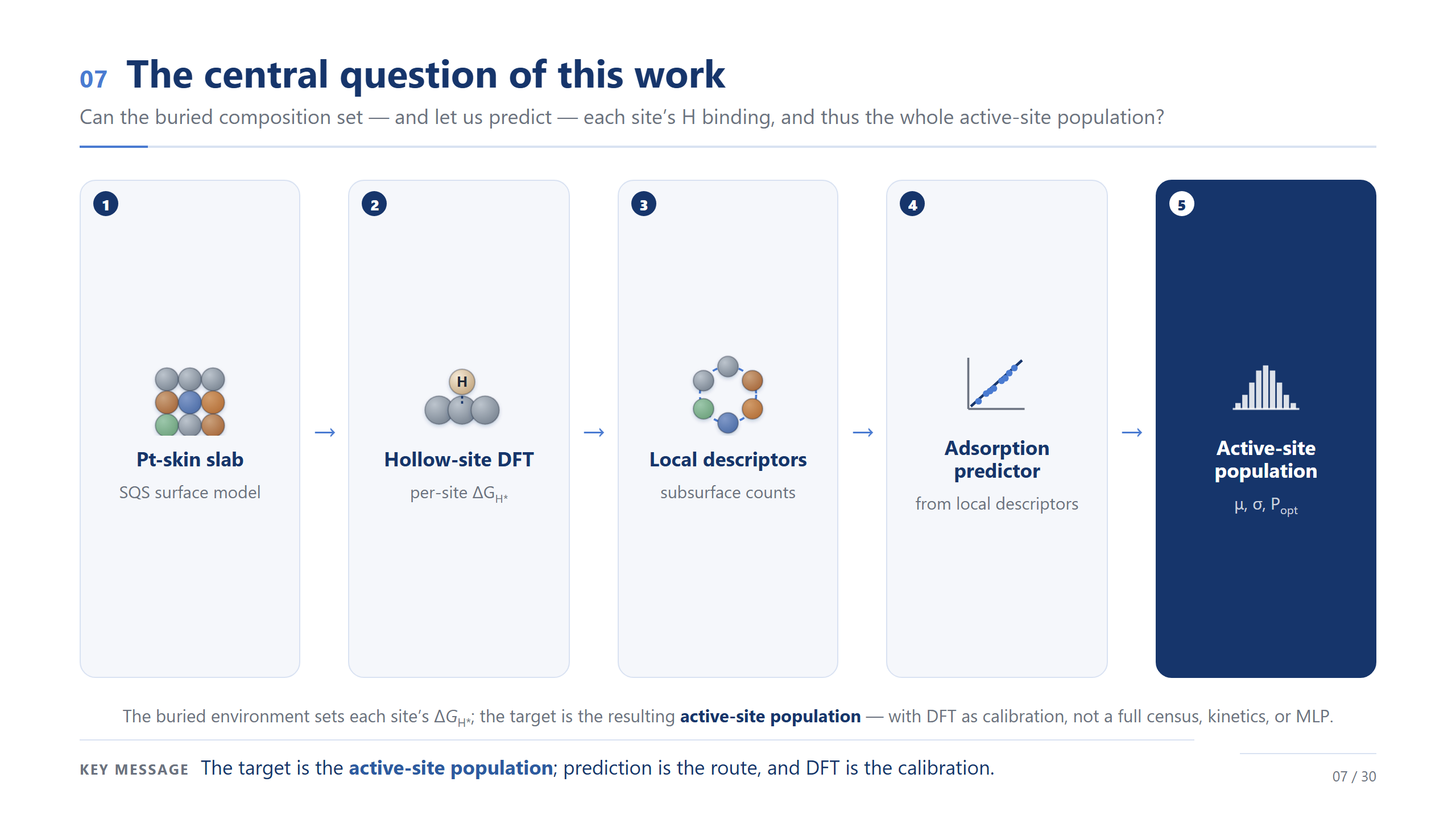
核心問題:預測族群
於是核心問題就很清楚了:底下的成分,能不能決定、進而讓我們預測每個位點的氫吸附,然後拼出整個族群?如果可以,DFT 的角色就變了——它不再是把每個位點都算一遍的普查,而是少數幾個位點的校準資料。請看這條流程的終點:不是預測器,而是「活性位點族群」。預測是路,族群才是目的。

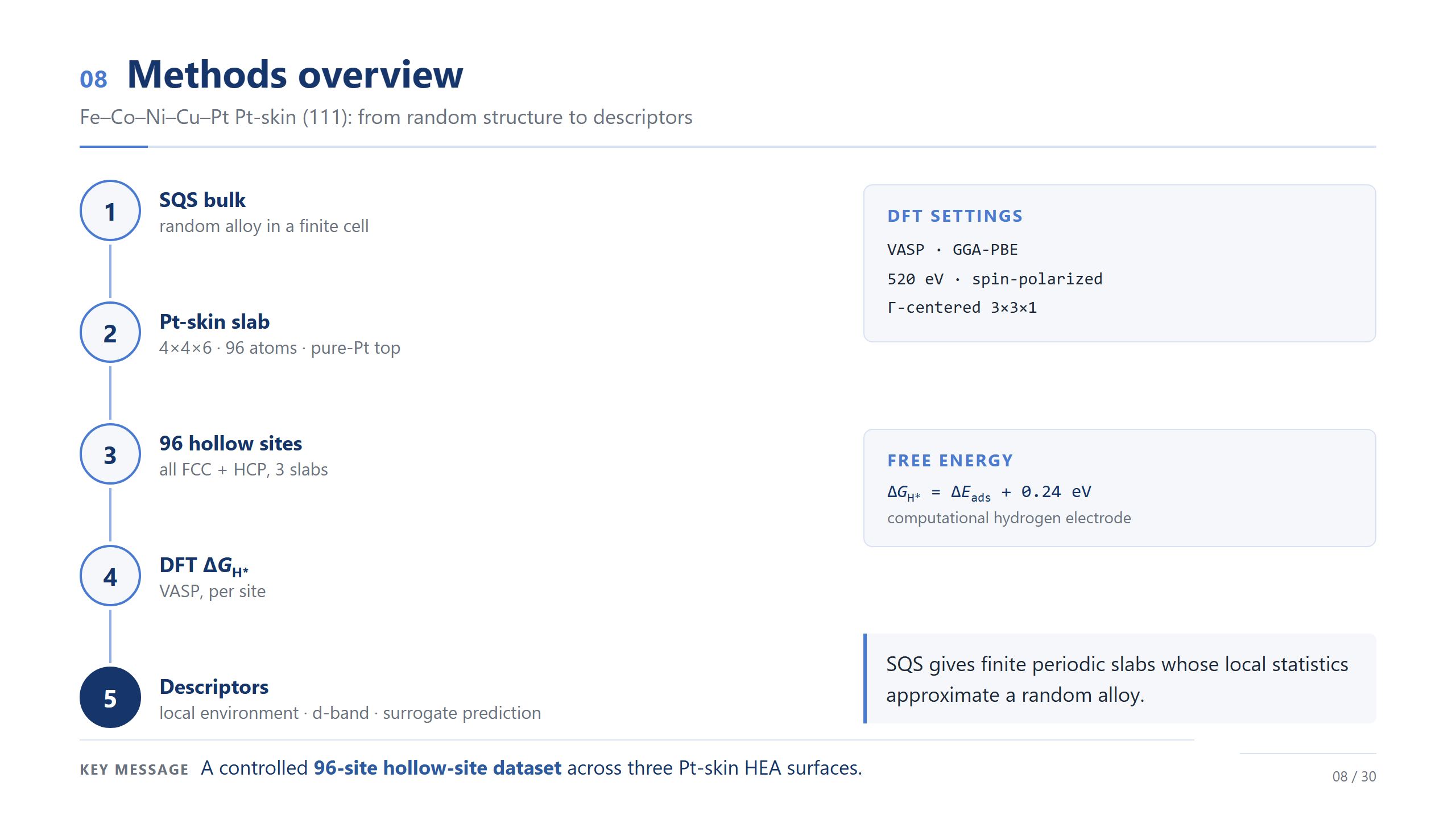
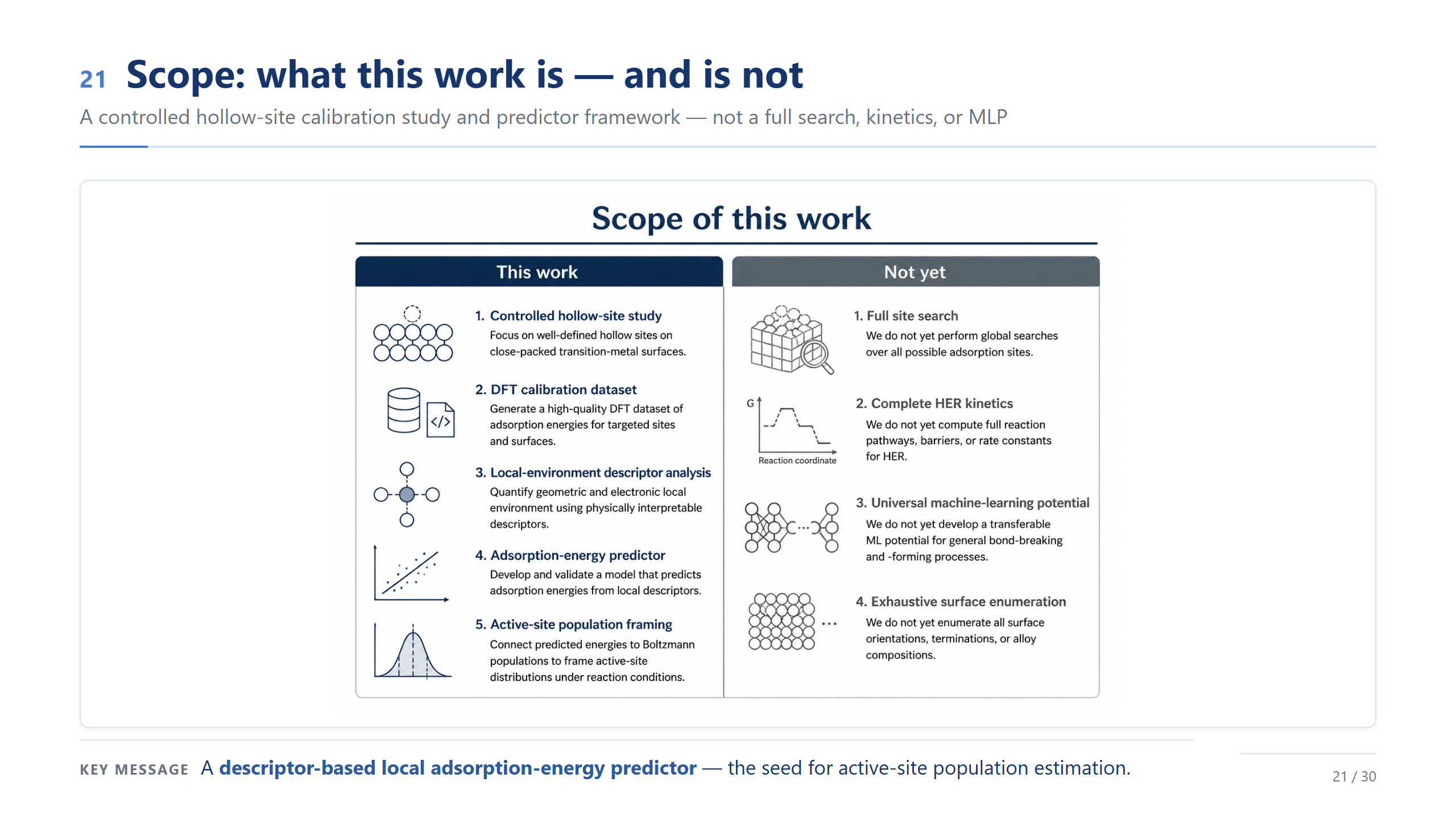
方法總覽(精簡)
方法我講最精簡的版本。我們做 Fe–Co–Ni–Cu–Pt 的 Pt-skin (111):用 SQS 生一個統計上接近隨機的合金、切成 96 原子的 slab、頂層換純 Pt、在所有 hollow site 算 ΔG_H*、再抽局部描述符;設定是 PBE、520 eV、自旋極化。各位只要記得:這是跨三個 Pt-skin 表面、96 個 hollow site 的一份受控資料集——它是用來校準的,不是終點。
小胞能代表無限大隨機合金嗎?
這裡花一頁解釋我們為什麼需要 SQS。一個真正的隨機合金有大約 10 的 23 次方個原子,DFT 一次卻只能算大約一百個。左邊是真實的隨機合金,右邊是我們算得動的超胞,中間這個問號就是難點:能不能用一個這麼小的胞,代表一個幾乎無限大的隨機合金?SQS 就是來填這個鴻溝的。
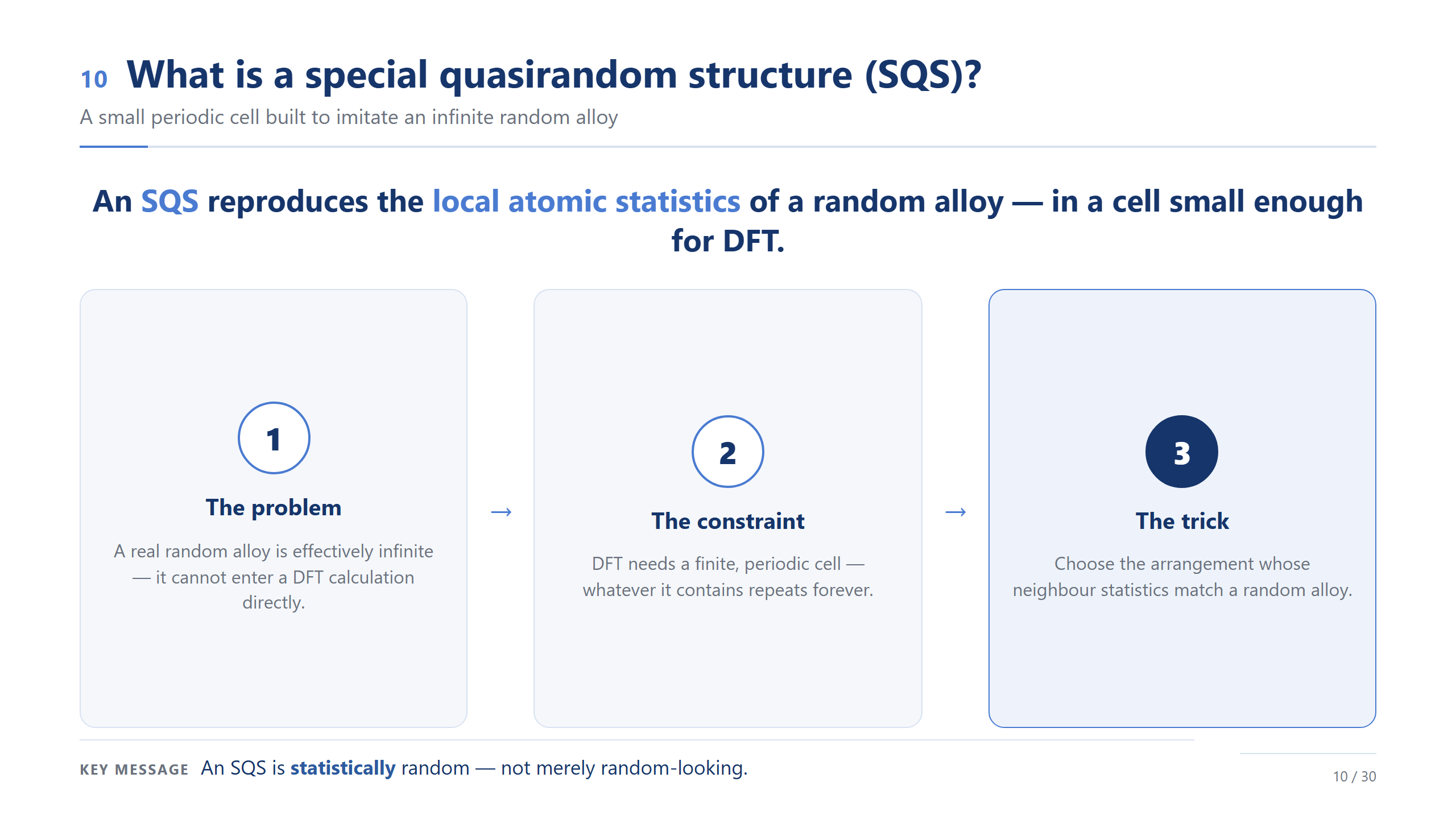
什麼是 SQS
SQS 的答案很聰明:不要求胞很大,只要求它的「鄰居統計」跟隨機合金一樣。所以它的隨機是統計意義上的隨機,不是看起來亂就算數。主流程我只講到這——「我們有一個小胞,統計上等於隨機合金」;它具體怎麼被找出來,我放在最後的 backup,有興趣再看。各位只要相信:這個 96 原子的模型,統計上代表一個隨機的高熵合金。

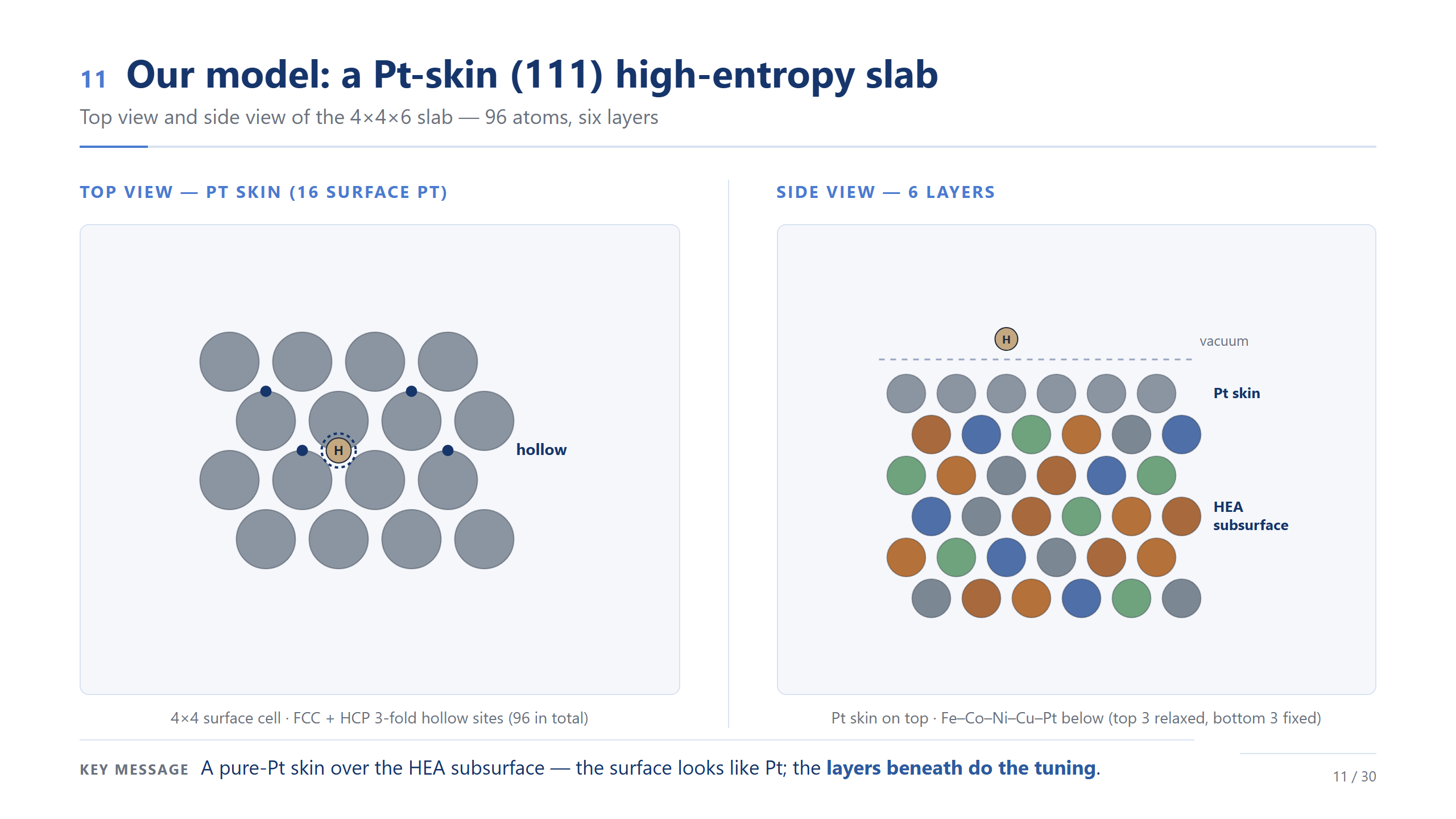
我們的模型:Pt-skin (111) slab
這就是我們的模型。左邊俯視:表面 4×4、16 個 Pt 的 skin,氫吸在這些三配位 hollow site 上;右邊側視:六層,最上一層純 Pt,底下五層才是 Fe–Co–Ni–Cu–Pt 的混合,計算時放鬆上三層、固定下三層。一句話:表面看起來就是 Pt,真正在做調控的,是底下那幾層。
數什麼:hollow 底下六顆原子
那我們到底數什麼?把 Pt skin 想成透明的往下看:每個 hollow 正下方剛好坐著六個 subsurface 原子,這就是它的局部環。我們最主要的描述符 ls_X,就是這六個原子裡各元素各有幾個。所以每個位點的身分證,就是它 hollow 底下那六顆原子的組成——非常簡單、可數。
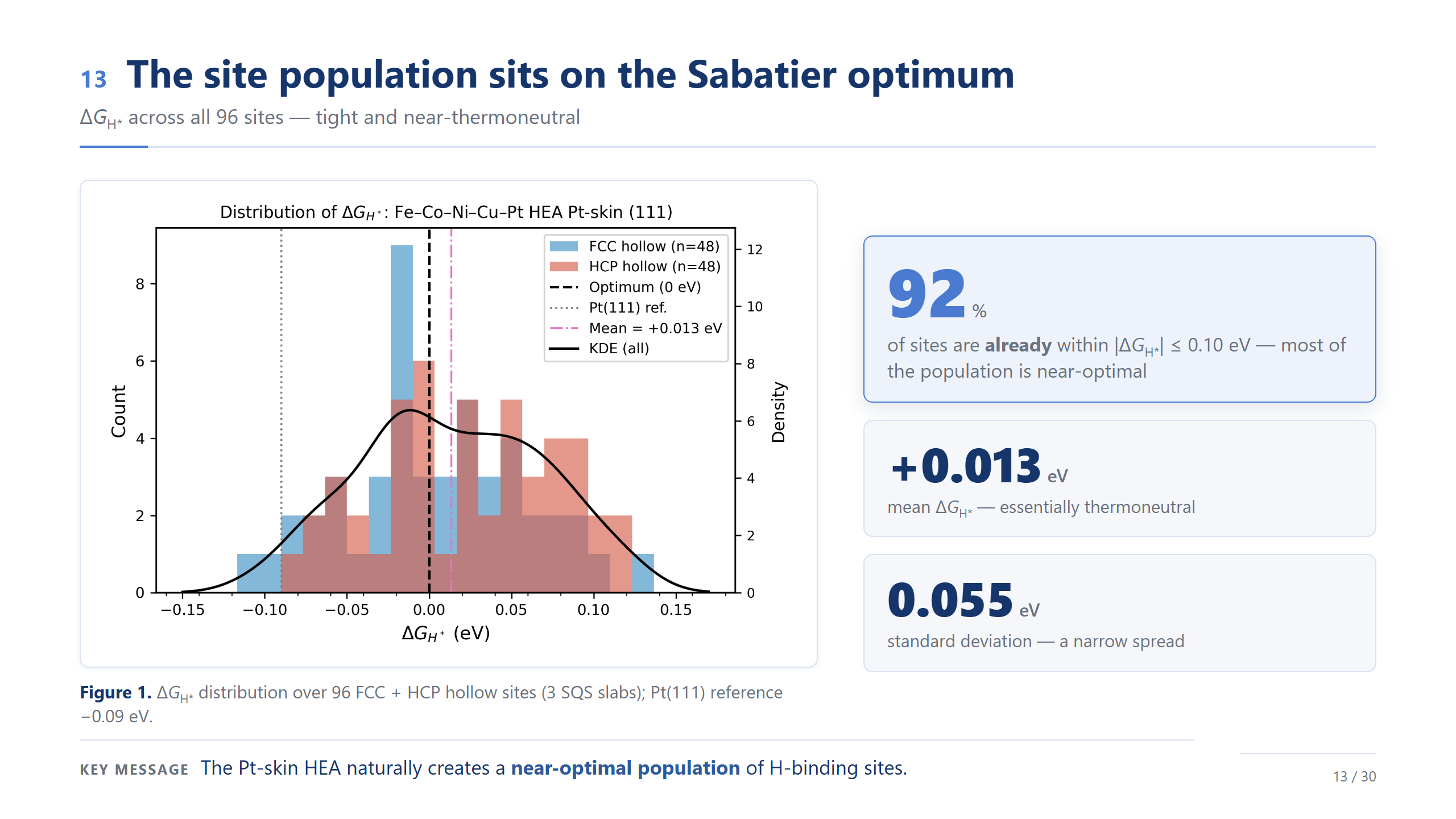
位點族群落在 Sabatier 最佳點(92%)
先看結果,這也是今天第一個漂亮的數字:96 個位點裡,有 92% 的 ΔG_H* 絕對值「已經」落在 0.10 eV 以內。平均 +0.013 eV、幾乎熱中性,標準差只有 0.055 eV、非常窄;對照純 Pt(111) 是 −0.09 eV。換句話說,這個 Pt-skin 高熵合金,自己就長出了一整個接近最佳的位點族群。
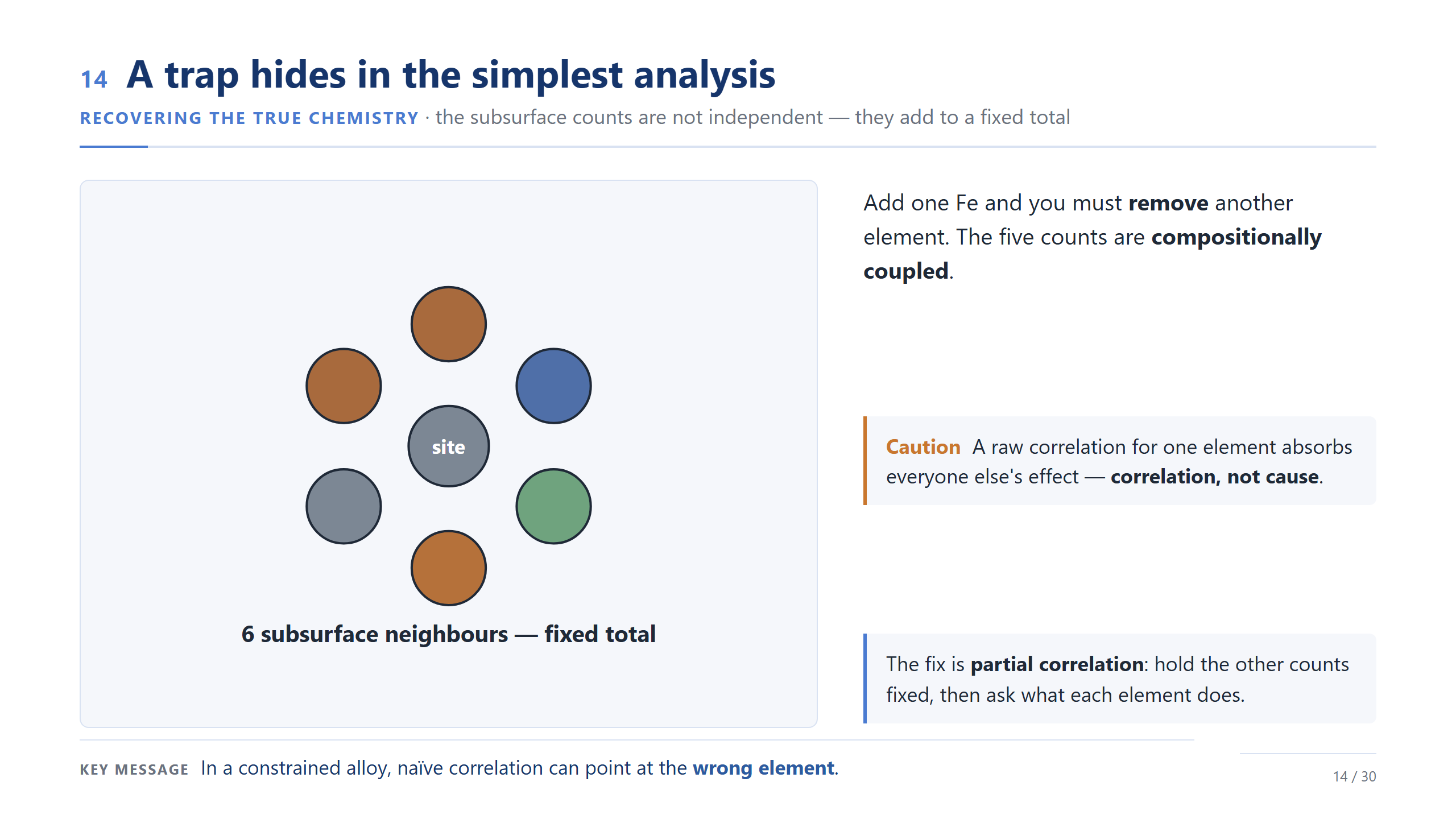
局部描述符受成分封閉約束
在解讀相關性之前,有一個重要的細節。每個 hollow site 是用它底下的六顆次表層原子來描述的,所以五種元素的個數永遠加起來等於六——Fe 多一個,至少就有另一種元素少一個。因為這種成分封閉,raw Pearson 相關當初步篩選很有用,但不能直接當成某個元素的獨立效應。
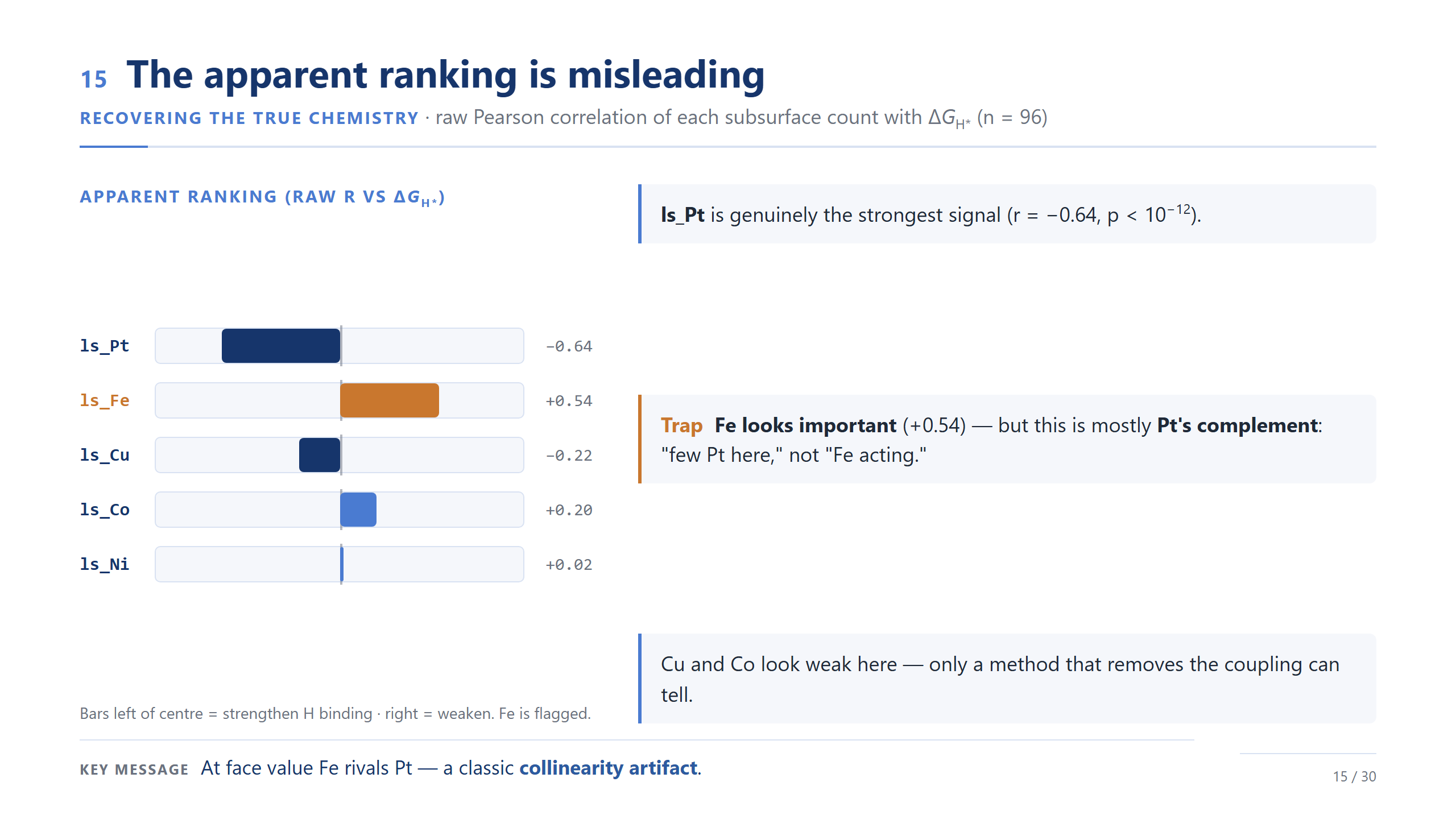
raw Pearson:Pt-rich / Fe-rich 趨勢
raw Pearson 給了第一個線索:局部 Pt 多的環境,氫吸附偏強;局部 Fe 多的環境,氫吸附偏弱。但因為這些 count 是封閉的,我們先不急著說這是 Fe 的獨立效應。這只是一個「表面上的排序」,下一步要做一個成分上的檢查。
Fe 只是 Pt 的補集嗎?
這裡我想排除一個簡單的替代解釋。因為描述符加起來是六,Fe 多的位點也許只是 Pt 少的位點。但資料不支持這個說法:Pt 跟 Fe 甚至不是最強的反相關對,Pt–Ni 比 Pt–Fe 還反相關;更重要的是,當我們比較「Pt 數目相同」的位點時,Fe 跟 ΔG 仍然是正相關。所以 Fe 不只是 Pt 的反面,它是一個有意義的、偏弱鍵結端的次表層描述符。
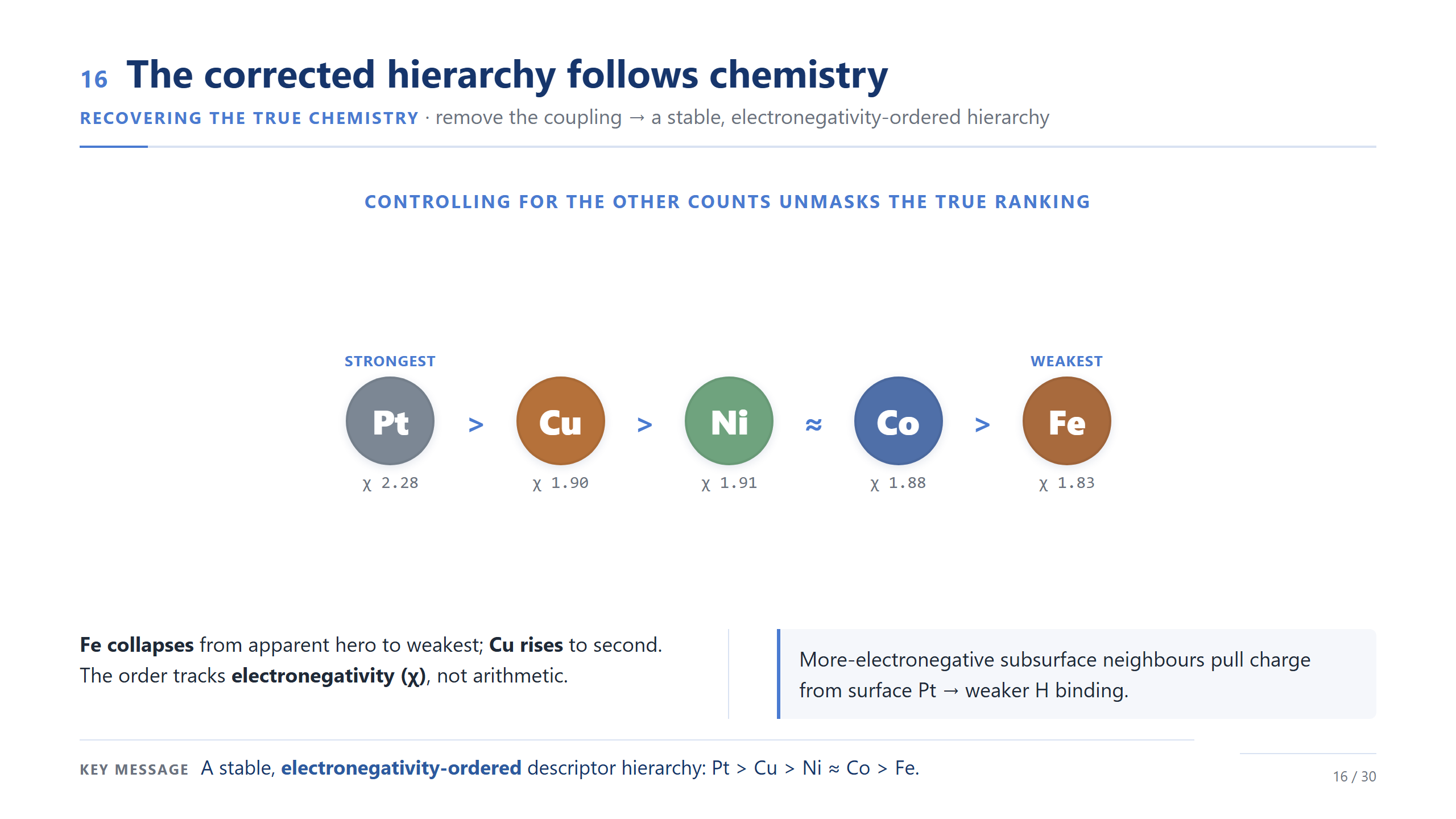
取代效應給出化學可讀的排序
因為描述符是封閉的 count,更自然的解讀,不是去問每個元素的絕對效應,而是問:把一顆次表層原子換成另一種會怎樣。這個取代效應給出一個化學上好讀的趨勢:Pt 讓鍵結最強、Fe 最弱,Cu、Ni、Co 在中間;把一顆 Fe 換成 Pt,鍵結大約強 61 meV。這個趨勢跟電負度順序一致,不過我會把它當成化學上的一致性,而不是單獨的證明,而且只在這份資料集內成立。
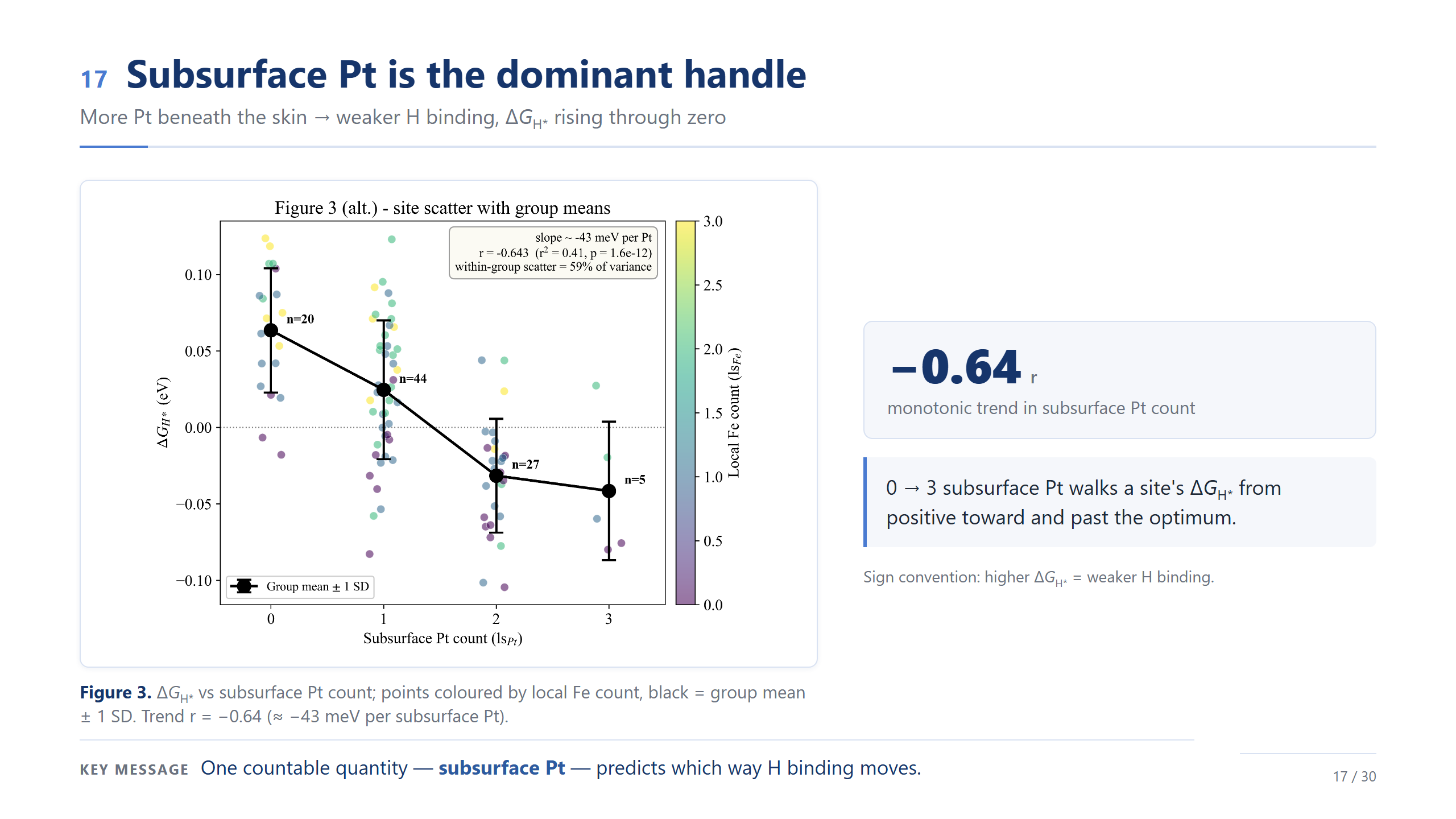
subsurface Pt 是主導旋鈕
在這個排序裡,最主導的單一旋鈕,就是 subsurface Pt 的數量。橫軸是底下 Pt 個數、縱軸 ΔG_H*,相關 −0.64、大約每多一個 Pt 變化 −43 meV;底下 Pt 從 0 到 3,把位點一路帶過零點。但注意黑色的群組平均——趨勢雖明確,組內散布還是很大,這再次提醒我們它是一個族群。一句話:光是底下有幾個 Pt,就能預測氫吸附往哪走。
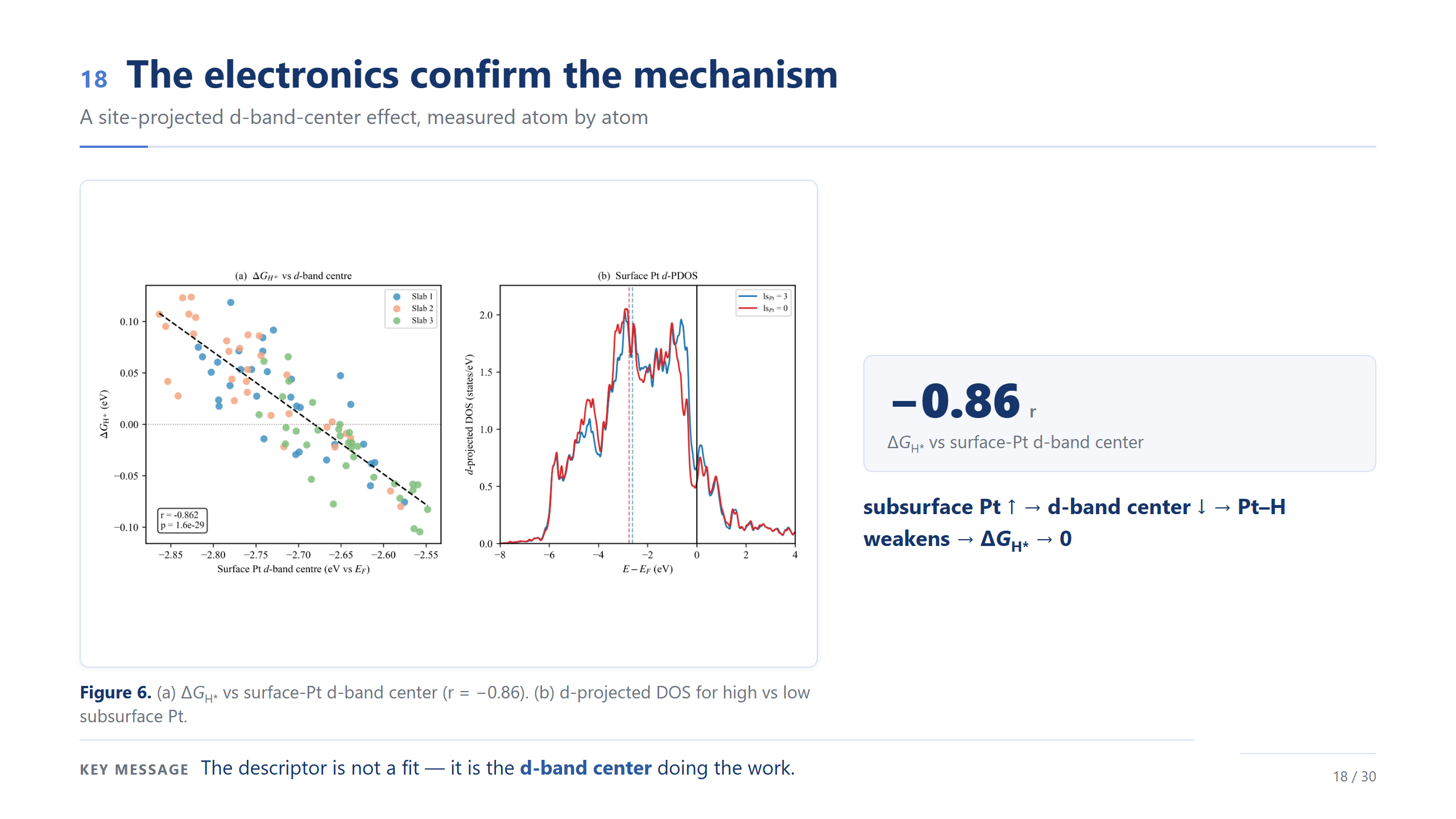
電子結構印證機制(d-band)
那背後的物理是什麼?是 d-band center。我們一顆一顆抽表面 Pt 的 d-band center,跟 ΔG_H* 的相關高達 −0.86;右邊 PDOS 也看得到,底下 Pt 多的時候,表面 Pt 的 d 態整個下移。所以 Pt 這一端的趨勢真正在背後做功的,是 d-band center;至於 Fe 端的微觀機制,還需要進一步的電子結構驗證,我會講得保守一點。
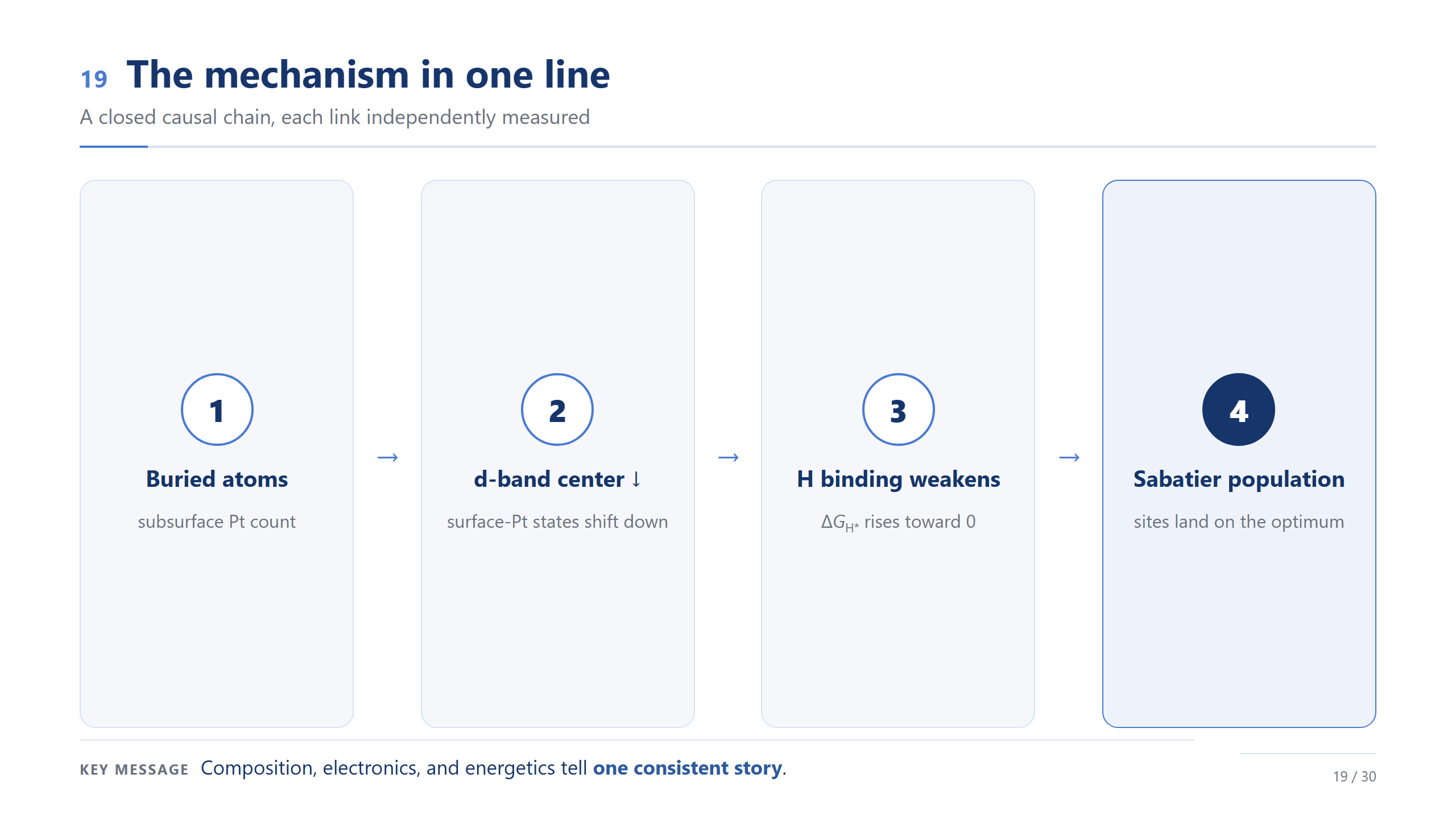
一行講完機制
把 Pt 這一端的因果鏈串成一句:底下 Pt 上升、表面 Pt 的 d-band center 下移、Pt–H 變弱、ΔG_H* 往零靠近,最後所有位點落在最佳點附近、形成族群。成分、電子、能量,在 Pt 這一端講的是同一個一致的故事。
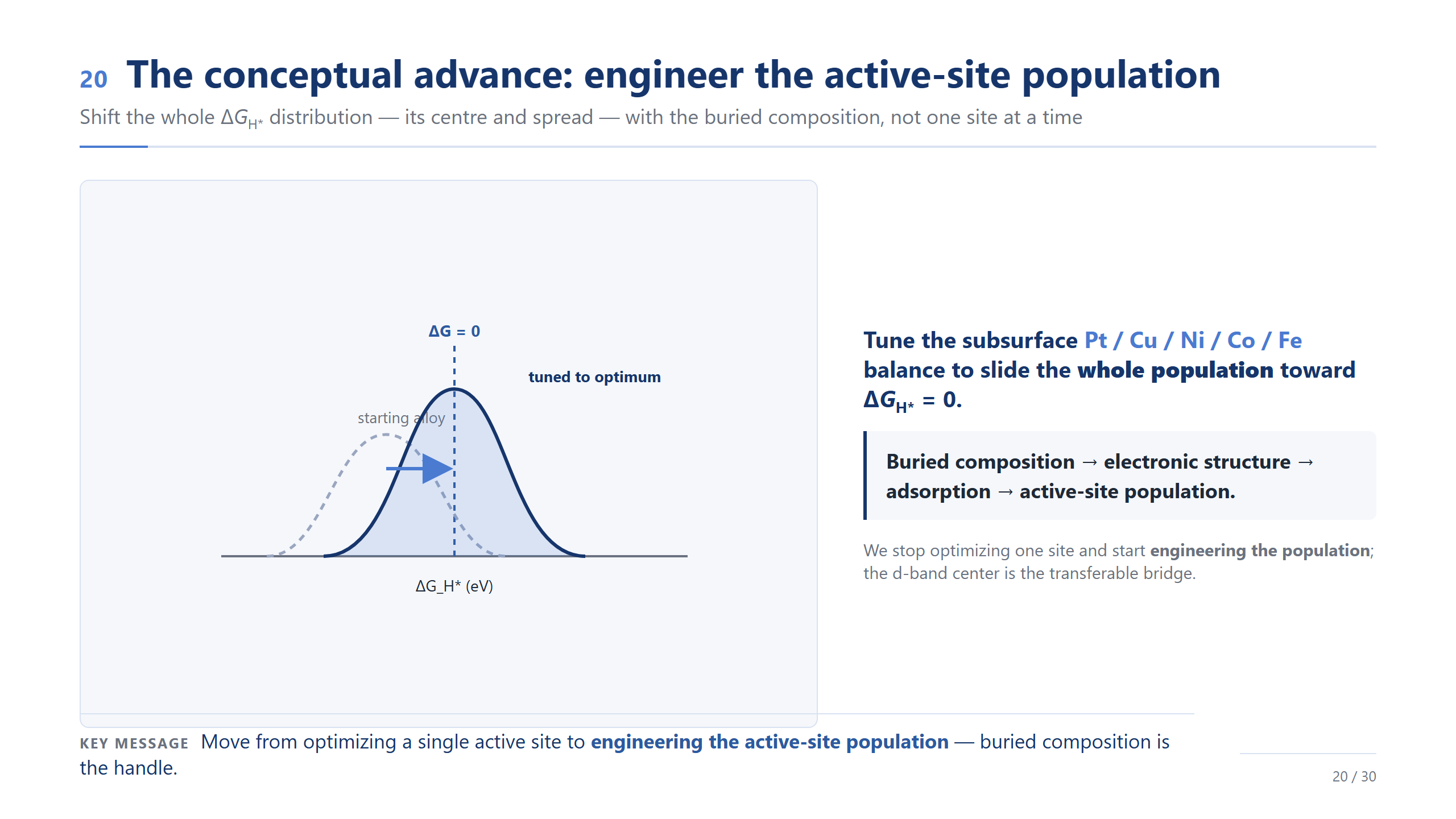
觀念進展:設計位點族群
如果整場只記一頁,我希望是這一頁——這是這份工作真正的觀念進展。設計的對象,應該是整個 ΔG_H* 分布,它的中心和寬度,而不是某一個位點。請把這條鏈記起來:埋藏成分 → 電子結構 → 吸附 → 活性位點族群。我們不再優化一個位點,而是開始設計整個族群,而底下的成分,就是那個旋鈕。

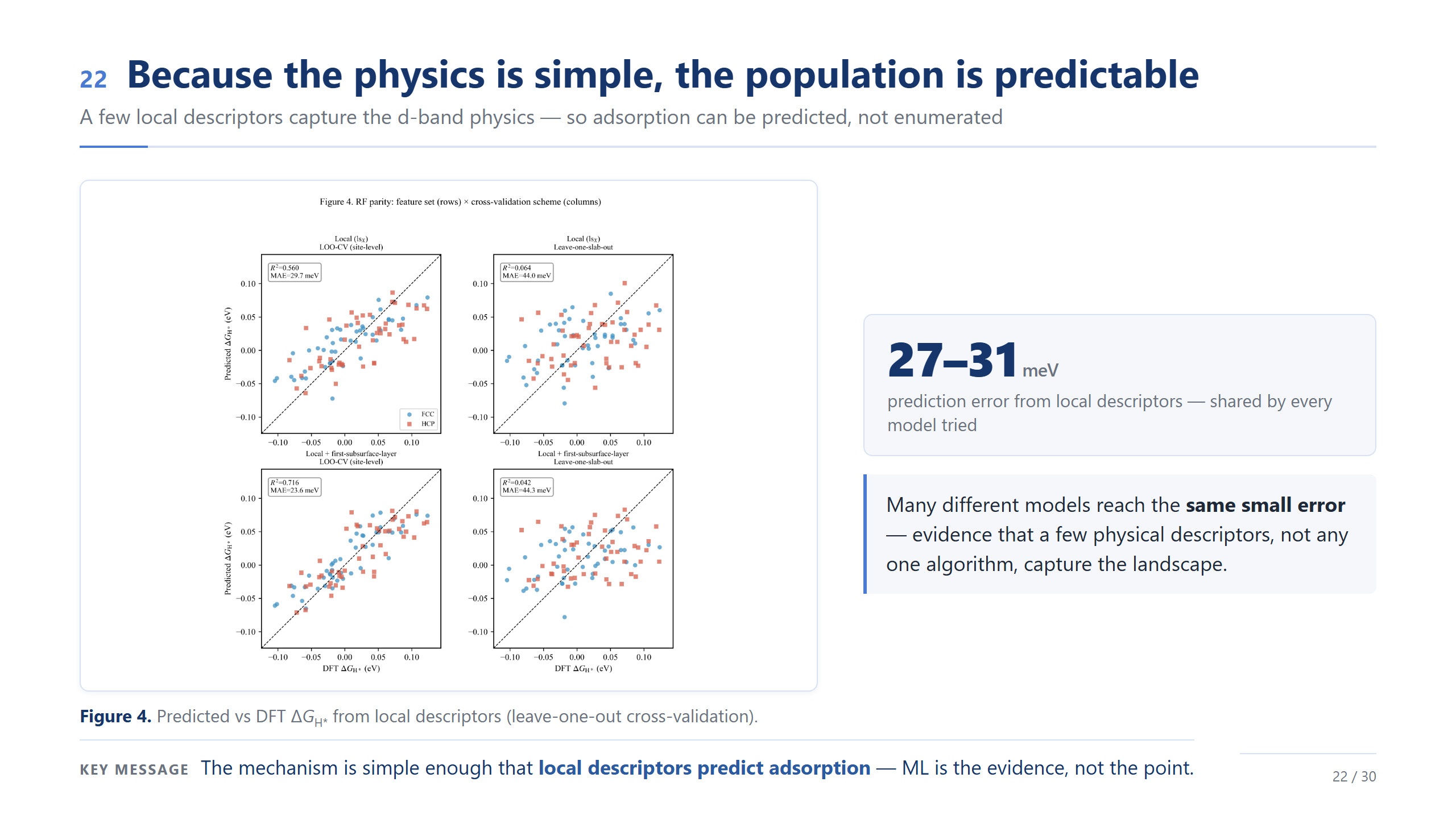
物理簡單,所以族群可預測
來看機器學習的驗證,這也順便幫我們定出這份工作的邊界。在 site-level 的 leave-one-out 下,只用幾個局部描述符,誤差大約落在 24 到 30 meV,代表描述符確實抓到了主要趨勢。但請特別看右邊那兩格:換成 leave-one-slab-out、也就是要去預測一個沒看過的 slab 時,模型幾乎崩掉。所以這裡的訊息不是「我們做出了一個 predictor」,而是:機器學習幫我們界定範圍——它適合在已取樣的 slab 裡做描述符的解讀,不能外推到沒見過的成分。這個 validation 結果也幫我們定義了這篇工作的邊界:我們不把這個模型宣稱成可以外推到新成分的 predictor,它的角色是幫助我們在已取樣的 Pt-skin HEA slabs 裡,理解 local environment 如何控制 ΔG_H*,以及這些 site-level 的 ΔG_H* 如何形成 active-site population。所以最後我用三點總結今天的主軸。

結論 · Conclusion
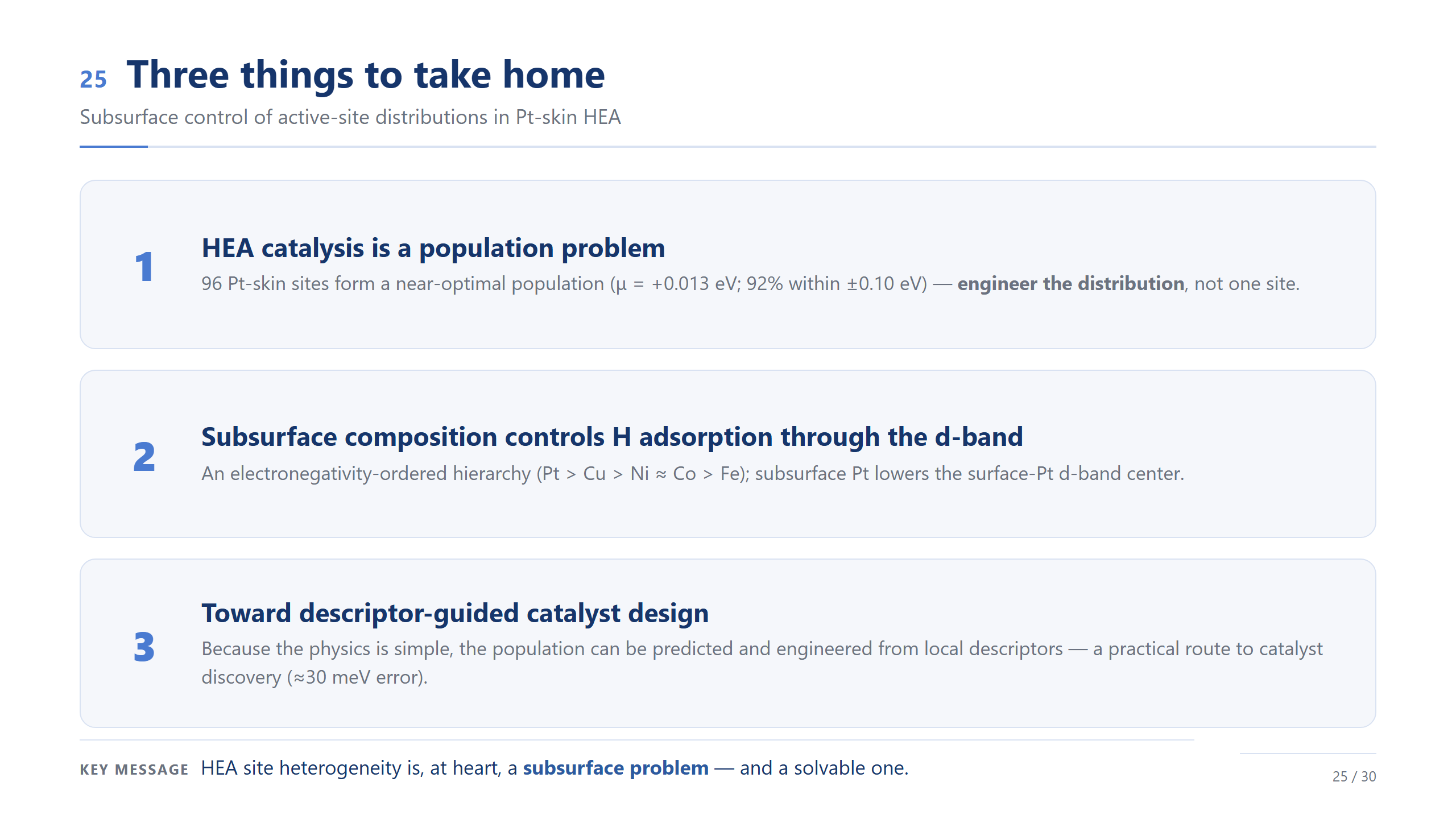
最後總結三點。第一,高熵合金催化不是只看單一最佳位點,而是一個 population problem——真正要設計的是整個 ΔG_H* 分布,以及接近 optimal 的 active-site 比例。第二,在這個 Pt-skin HEA dataset 裡,subsurface composition 會透過 d-band 影響 H adsorption;也就是說,底下那一層原子的組成不是背景,而是一個可以調控表面 Pt 活性的 handle,在這份資料中,我們看到不同 subsurface elements 對 H binding 有不同方向的影響。第三,這個結果提供 descriptor-guided population design 的起點,但我們也很清楚,前面的 ML validation 顯示它目前還不是可以外推到新成分的 predictor;所以目前最安全的 claim 是:在已取樣的 Pt-skin HEA slabs 中,我們可以用 descriptor 來理解 local environment、ΔG_H* distribution,以及 active-site population 之間的關係。下一步才是擴大到更多 slabs 和更多 compositions,建立真正的跨成分模型。
結語 · Thank you
總結成一句話,也是我今天最想留給各位的:設計那群埋在底下的原子,而不只是表面的活性位點。埋藏成分 → 電子結構 → 吸附 → 活性位點族群,這就是 Pt-skin 高熵合金的設計框架。謝謝大家,也歡迎指教;這份工作感謝 NSTC 的支持與我的學生們。

Backup · SQS 怎麼建
以下是 backup,留給 Q&A。如果有人問 SQS 到底怎麼被找出來,我們可以翻到這裡:怎麼挑排列、以及用蒙地卡羅退火怎麼找到它。

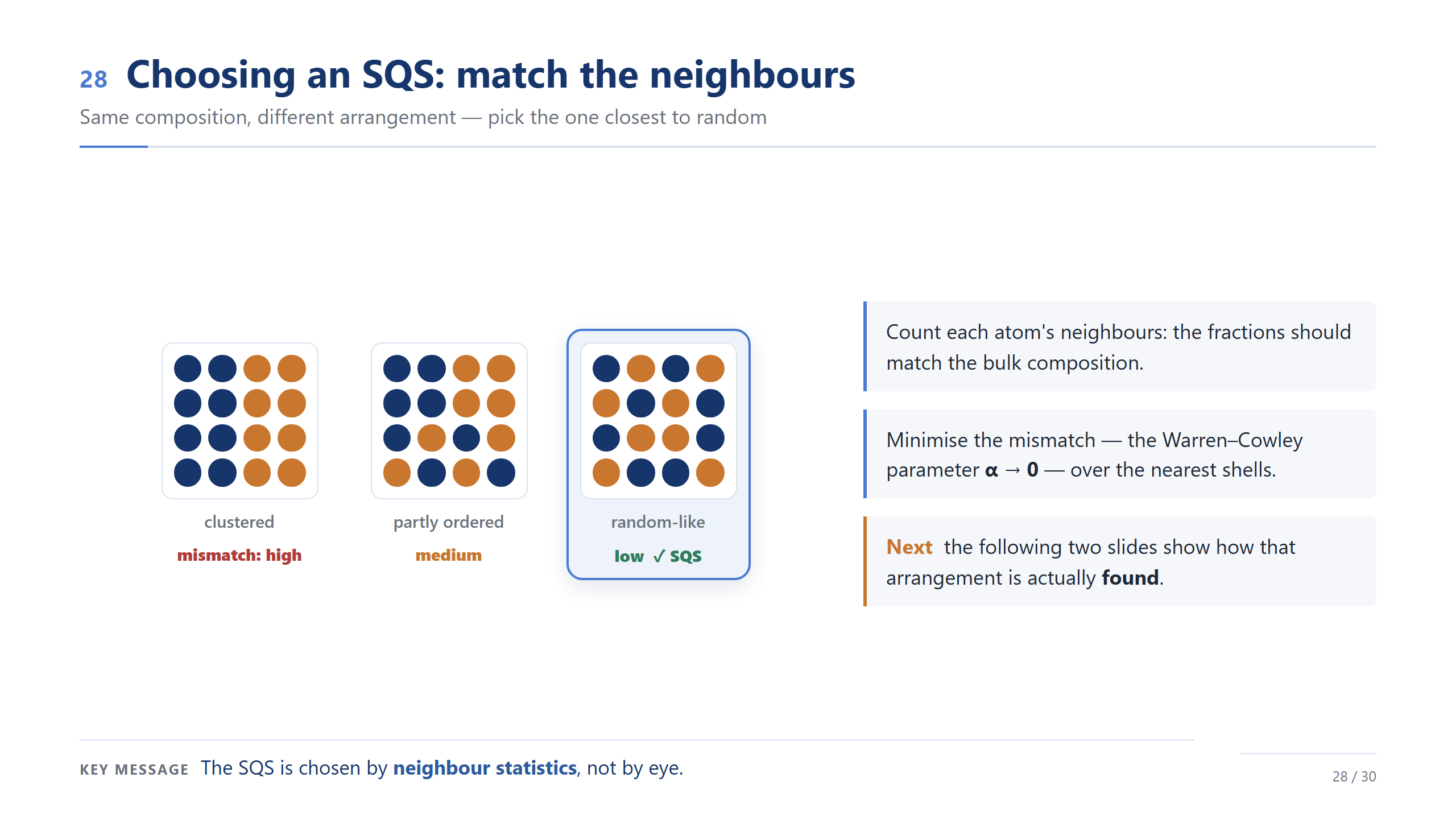
怎麼挑 SQS(backup)
挑 SQS 的關鍵是看鄰居。同樣成分有很多排法:團簇的跟隨機差很遠,部分有序的好一點,鄰居統計最接近隨機的那個才是 SQS。我們用 Warren–Cowley 參數量化落差、讓它趨近零。重點:SQS 是用鄰居統計挑的,不是用眼睛看的。
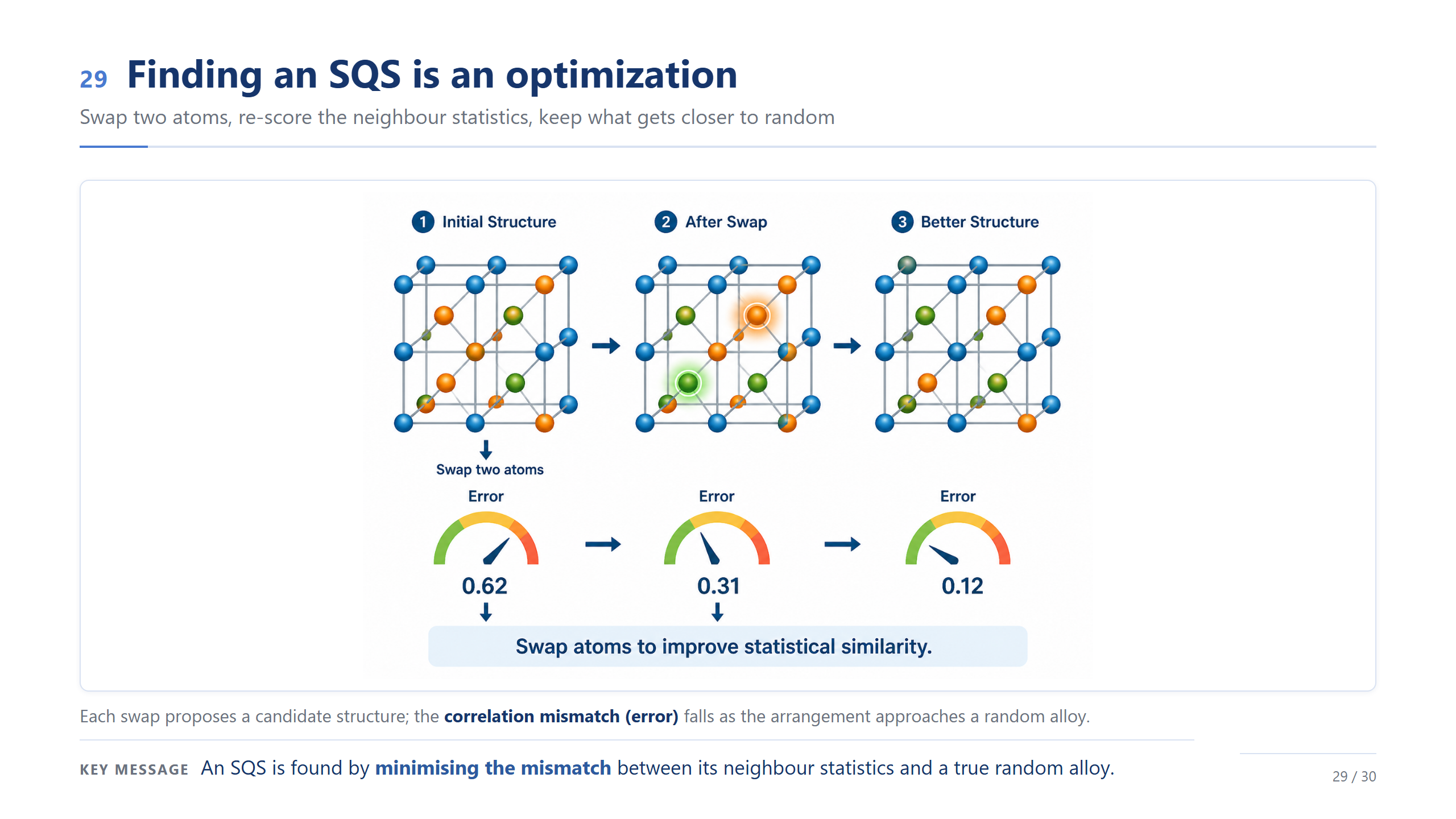
找 SQS 是最佳化(backup)
而「挑」本身就是一個最佳化。隨機交換兩個原子、重算鄰居統計的誤差,變好就保留——你看誤差從 0.62、0.31 一路降到 0.12。找 SQS,就是最小化我的結構跟真隨機合金之間的統計落差。
蒙地卡羅退火(backup)
但只接受變好的交換會卡在 local minimum。所以我們用蒙地卡羅退火:變好一定接受,變差也以 exp(−ΔE/T) 的機率接受,溫度再慢慢降;這樣才能找到統計誤差全域最低的結構,而不是停在附近的小坑。這就是我們產生 SQS 的方式。